Unlock Your Potential: The Ultimate Guide to Genome Sequencing

Genome Sequencing: The Definitive Guide From Sample to Insight—With Real-World Wisdom
Genome sequencing isn’t just a buzzword or a fancy lab technique—it’s a complex adventure filled with unexpected twists, surprises, and sometimes outright headaches. Over nearly a decade of experience, I’ve learned that mastering genome sequencing isn’t about blindly following protocols; it’s about anticipating problems before they hit, troubleshooting like a detective, and understanding the biological story buried beneath mountains of data.

This guide is my honest, no-fluff account of genome sequencing—from sample collection to final analysis—with practical tips, real mistakes I’ve made (so you don’t have to), and actionable next steps. Whether you’re stepping into this field for the first time or refining your existing skills, consider this your trusted companion.
1. Genome Sequencing: More Than Data—It’s a Puzzle You Have to Solve
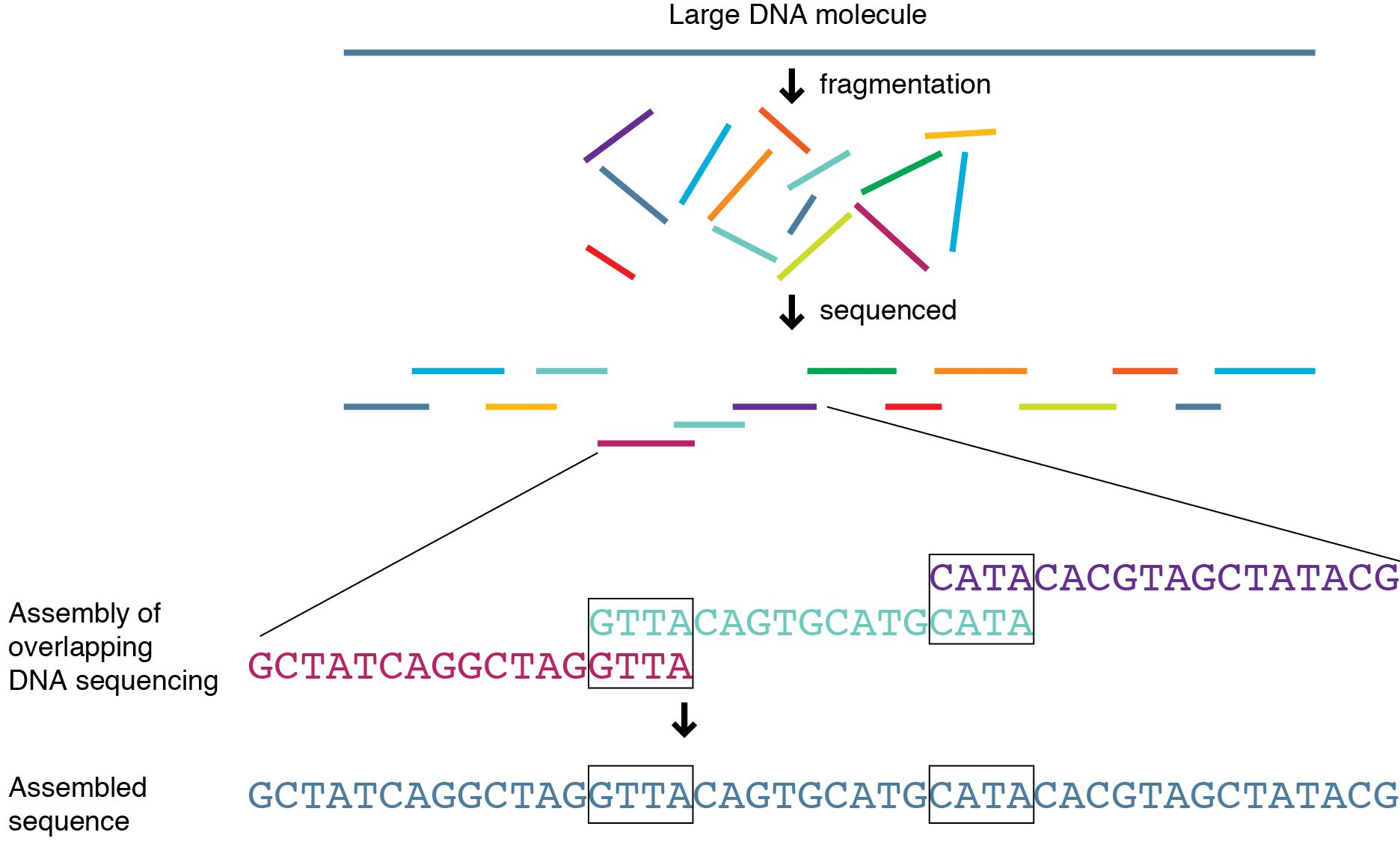
At its core, genome sequencing turns DNA’s four-letter code (A, T, C, G) into digital information. But here’s the catch: DNA doesn’t arrive neatly packaged. It’s more like shredded pages from dozens of books mixed in one box. Your job is to piece those pages back together into meaningful stories.
When I started in 2015 at a small environmental microbiology startup, I thought running the sequencer was the hardest part. Nope. The real challenge? Sample prep and interpreting results. Each step can introduce errors—contaminated samples, degraded DNA, or misreads—that snowball if you don’t catch them early.
Key takeaway: Don’t treat sequencing as “just data generation.” Think of it as forensic reconstruction where every tiny detail matters.
2. Sample Collection: The Foundation That Makes or Breaks Everything
I still cringe remembering when a conservation group handed me plant leaves stored loosely in plastic bags at room temperature for three days before extraction. Result? Completely degraded DNA and $5,000 down the drain.
Here’s what often goes wrong—and how to fix it:
| Problem | Why It Happens | How To Fix It |
|---|---|---|
| DNA degradation | Warm storage delays; nuclease activity | Flash freeze samples immediately (liquid nitrogen preferred) or use preservation buffers |
| Contamination | Microbial growth; dirt; chemicals | Collect with gloves; sterilize tools; use clean tubes |
| Poor purity | Co-extracted proteins or phenols | Use commercial kits like Qiagen DNeasy Plant Mini; verify purity ratios on Nanodrop (260/280 = 1.8–2.0) |
| Low integrity | Mechanical damage during handling | Handle gently; avoid repeated freeze-thaw cycles; confirm via gel electrophoresis |
Pro tip: Less DNA but high-quality beats loads of junk every time. I’ve seen projects saved simply by discarding degraded samples—even if quantity was low.
3. Choosing Your Sequencing Platform: Match Tech to Your Goal
There’s no magic bullet platform—your choice depends on your project’s unique needs.
Here’s what years of trial and error taught me:
| Use Case | Recommended Platform | Why |
|---|---|---|
| Complex microbial communities | Oxford Nanopore MinION | Long reads untangle repeats & unknown species despite higher error rates |
| Human exome/genome sequencing | Illumina NextSeq/HiSeq | High accuracy & mature pipelines for clinical-grade data |
| Rapid outbreak response | Oxford Nanopore MinION | Portability & near real-time data streaming |
Caution: Don’t pick based on hype alone! Define your questions first—accuracy vs read length vs turnaround time—and then choose accordingly.
4. Library Preparation: Avoiding Nightmares That Waste Weeks
Library prep is where many good projects start dying slowly—and silently.
Common traps I still trip over occasionally:
- Fragmentation inconsistency: Covaris sonicators need regular calibration; otherwise fragment sizes vary wildly and mess up downstream assembly.
- Adapter dimers: Skipping bead cleanup once led me to >30% adapter contamination—a nightmare in data filtering.
- Cross-contamination: Pipetting multiple samples without changing tips caused barcode collisions that took days to diagnose.
How I fixed these issues:
- Regular calibration logs and SOP checklists.
- Always using AMPure XP beads post-ligation.
- Physically separating pre-PCR (clean) from post-PCR areas.
If budget permits, automating library prep with robots like Agilent Bravo can reduce human error dramatically—I saw failed preps drop by over 60% after automation in my last lab stint.
5. Running the Sequencer: Patience Is More Than Virtue—It’s Survival
One tough lesson I learned early on: never underestimate run monitoring.
Once an Illumina NovaSeq run failed halfway because fluidics clogged—hours later we realized we lost over 600 GB of raw data that could never be recovered.
What keeps runs running smoothly?
- Live monitoring using Illumina SAV software—or equivalent for other platforms—to catch drops in quality metrics ASAP.
- Scheduling maintenance strictly based on run counts rather than calendar dates.
- Avoid starting critical runs right before weekends or holidays when no one can intervene if something goes wrong.
Bonus tip: For questionable samples, always run a small pilot lane first—it might feel like extra work but saves you from catastrophic failures on full runs down the line.
6. Bioinformatics: Navigating the Maze Without Getting Lost
Bioinformatics can feel overwhelming—the raw FASTQ files look like random gibberish until pipelines turn them into biological stories. Yet many stumble here due to subtle pitfalls:
| Issue | Cause | How To Fix |
|---|---|---|
| Low-quality reads | Bad sample prep or sequencer glitches | Run FastQC + TrimGalore for trimming poor bases |
| Misaligned reads | Wrong reference version/settings | Check reference genome accuracy; tweak BWA parameters |
| False positive variants | Low coverage or poor filters | Increase coverage (>30x recommended); apply GATK best practices filters |
| Fragmented assemblies | Repetitive regions + low depth | Supplement with long-read data; use hybrid assemblers (e.g., MaSuRCA) |
I once wasted weeks chasing phantom SNPs caused by low coverage in cancer genomics until switching from 10x to 40x coverage combined with PacBio HiFi reads clarified the mutation landscape completely—a true game-changer!
7. Data Management: Don’t Let Storage Become Your Achilles’ Heel
Sequencing generates terabytes fast—and losing data means losing months of work (been there).
My evolving strategy looks like this:
- Upload raw + processed data immediately to AWS S3 buckets with lifecycle policies archiving old files automatically.
- Maintain local NAS devices as working copies for fast access.
- Run checksum verification (e.g., md5sum) after every file transfer to confirm integrity.
- Document metadata meticulously using tools like LabKey Server tracking sample provenance and pipeline versions.
Surprise insight: Cloud storage may seem pricey upfront but prevents catastrophic loss and enables smooth collaboration far better than juggling external drives or local servers alone.
8. Troubleshooting Checklist: Your Emergency Toolkit When Things Go Sideways
Whenever something breaks (and it will), this checklist has saved me countless headaches:
| Symptom | Likely Cause | What To Do |
|---|---|---|
| No reads after sequencing | Library prep failure/instrument error | Check library QC profiles; rerun instrument diagnostics |
| Very low read quality scores | Degraded sample/contamination | Re-extract DNA with fresh tissue; validate purity ratios again |
| Unexpectedly fragmented assemblies | Insufficient coverage/repeats | Boost sequencing depth; add long-read sequences |
| Excess adapter contamination | Incomplete bead cleanup | Optimize bead cleanups; verify adapter removal via Bioanalyzer traces |
| Confusing variant calls inconsistent with phenotype | Alignment errors/contamination/pipeline bugs | Visualize alignments with IGV browser; cross-check variants using orthogonal methods |
Pro tip: Keep thorough lab notes logging reagent lots, incubation times, temperatures—when troubleshooting complex issues later, retracing exact steps often reveals overlooked errors instantly.
9. Real-Life Case Studies That Saved Time & Money
Let me share two quick examples that underscore why troubleshooting matters:
Case Study A: Rare Disease Clinic
Early runs showed zero usable mutations despite strong clinical suspicion—a red flag.
By applying my checklist systematically:
- Fresh blood samples replaced archived frozen ones.
- Switched from standard Illumina library prep kits to PCR-free kits reducing amplification bias.
- Coverage bumped from 20x up to 40x.
- Updated variant analysis pipelines incorporating latest population databases were applied.
Result? Identification of a novel pathogenic mutation within three weeks—directly influencing patient care decisions faster than expected.
Case Study B: Environmental Metagenomics Mix-up
In another project analyzing soil microbiomes, initial results were dominated by common bacterial contaminants rather than target species due to cross-contaminated reagents.
Solution?
- Switched reagent lots and enforced strict cleanroom protocols during extraction/library prep.
- Added negative controls at every step for contamination tracing.
Outcome? Cleaner datasets revealing true community diversity previously masked by noise—invaluable for conservation efforts underway.
10. Advanced Strategies & Techniques Worth Exploring
Once comfortable with basics, these techniques can elevate your game:
-
Hybrid Assemblies: Combine short-read accuracy (Illumina) with long-read contiguity (PacBio HiFi/Oxford Nanopore). Tools like MaSuRCA or Unicycler excel here.
-
Unique Molecular Identifiers (UMIs): Tag individual DNA molecules pre-amplification to identify PCR duplicates accurately—critical for rare variant detection in cancer genomics.
-
Adaptive Sampling (Nanopore): Real-time selective sequencing reduces costs by enriching target regions on-the-fly without extra library steps.
-
Cloud-Based Pipelines: Platforms such as Terra.bio offer scalable workflows integrating best-practice tools without requiring massive local compute infrastructure.
11. Beginner-Friendly Primer: Glossary & Quick Start Checklist
Before diving deep, here are some key terms explained simply:
-
FASTQ File: Raw sequence file containing DNA reads plus quality scores per base.
-
Coverage: Average number of times each base is sequenced; higher coverage means more confidence in calling variants (~30–40x standard for human genomes).
-
Library Prep: Process converting extracted DNA into fragments suitable for sequencers—including fragmentation, end-repair, adapter ligation.
-
Adapter Dimers: Small unwanted fragments formed when adapters ligate together without DNA insert—interfere with sequencing efficiency.
Quick Start Checklist for New Projects
- Collect fresh samples using sterile technique; freeze immediately if possible.
- Extract DNA using validated kits appropriate for your sample type.
- Verify concentration and purity via Nanodrop/Qubit and gel electrophoresis.
- Choose sequencing platform based on your project goals.
- Prepare libraries following strict SOPs including bead cleanups.
- Run pilot sequencing lanes before full runs whenever possible.
- Monitor runs live and keep detailed notes at every step.
- Use established bioinformatics pipelines but inspect outputs carefully for anomalies.
12. Final Thoughts: Embrace Complexity With Confidence & Curiosity
Genome sequencing will test your patience more times than you’d expect—but that’s where mastery begins.
Each hiccup—from degraded samples to puzzling variants—is an opportunity disguised as frustration if you approach it methodically and keep learning along the way.
So yes—the process is messy and sometimes maddening—but persist through uncertainty armed with experience-backed strategies outlined here…and soon enough you’ll find yourself not just generating sequences but unlocking life-changing insights hidden inside those elegant codes of A/T/C/G letters.
Remember what kept me sane during my worst runs?
“Every glitch is just another piece falling into place.”
Dive in knowing that with patience, precision, and persistence—you’re not alone on this journey.
If you want my full spreadsheet detailing troubleshooting scenarios across hundreds of projects—or ready-to-use bioinformatics pipeline snippets tuned for robustness—just reach out anytime! Sharing hard-won lessons beats reinventing wheels hands-down.
Thanks for reading—feel free to ask any questions or share your own stories struggling through genome sequencing challenges!



