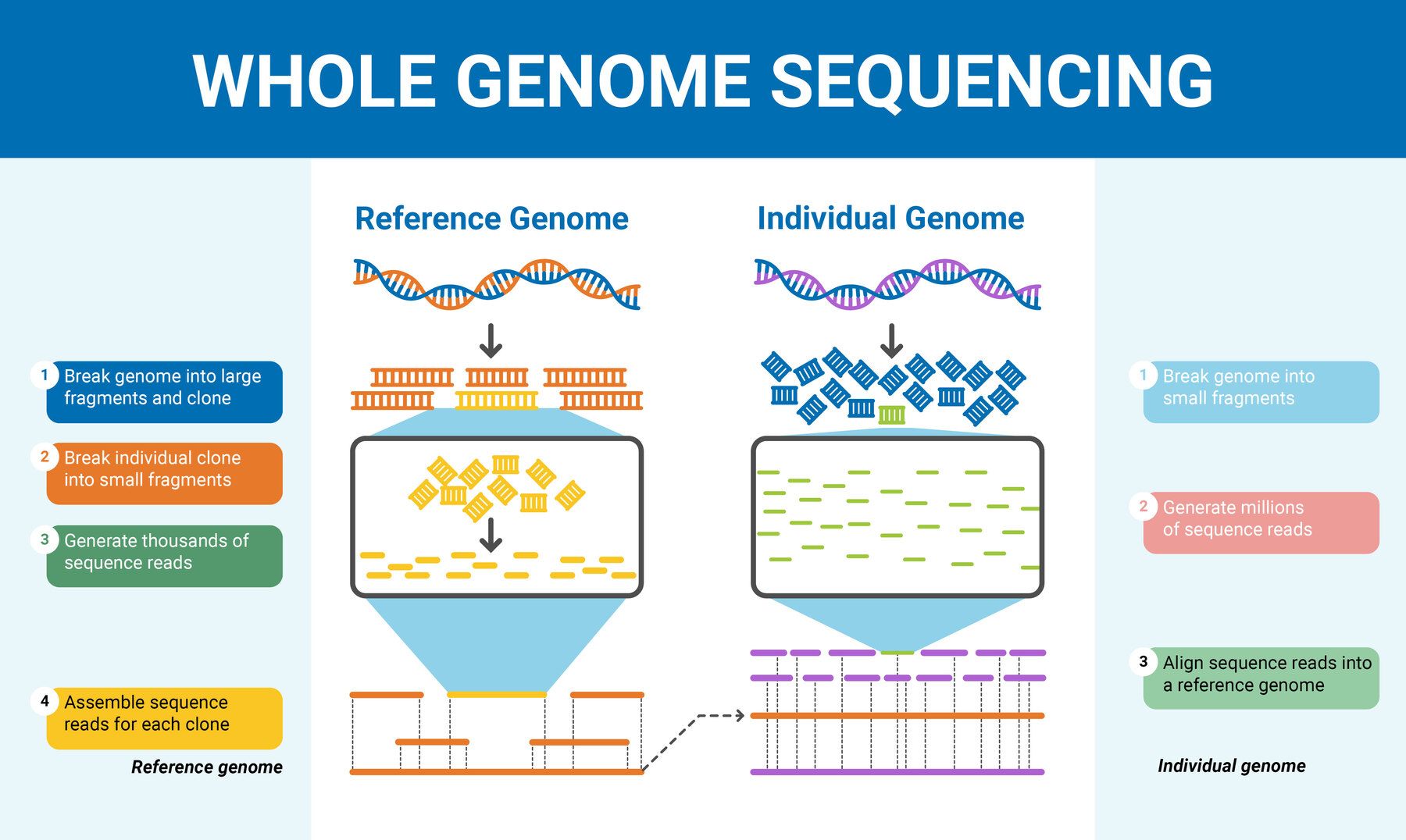
Understanding Genome Sequencing Technologies: Practical Types

When I first stepped into the world of genome sequencing during my grad school days, I was drowning in terminology and hype. Everyone acted like the newest tech was a magic wand that could solve every problem faster and cheaper. Spoiler: that’s dead wrong, and it took me a lot of trial and error to grasp why.

One big mistake that trips up nearly everyone—including me back then—is treating genome sequencing technologies as interchangeable upgrades rather than tools built for fundamentally different jobs. You’re not just choosing a faster way to read DNA—you’re picking a method with its own quirks, blind spots, and ideal use cases that dramatically affect your results.
Here’s a more personal, no-BS breakdown of what I learned after botching projects, getting stuck on data analysis, and finally understanding how to pick the right sequencing technology for the right question.
Why Sanger Sequencing Isn’t Dead Even Though Everyone Talks Like It Is
I’ll admit it: when I first encountered Sanger sequencing, I thought it was ancient tech best left in the past. But here’s the truth I learned after my first few projects: Sanger is still the go-to for accuracy when you need to nail down a specific mutation or verify results from more complicated methods.
Real talk: Sanger sequencing reads DNA fragments around 700–900 base pairs long by using chain-terminating nucleotides. Imagine carefully reading a book one short paragraph at a time to avoid any misread letters—that’s Sanger’s strength.
Example from my own lab: We had an NGS run showing a suspicious mutation in the BRCA1 gene of a patient sample. The data looked promising but also noisy. Instead of trusting it blindly, I sent that same region for Sanger sequencing. Boom—the mutation was confirmed cleanly within 48 hours, saving us weeks of chasing false leads.
Why this matters:
- Accuracy >99.99% means clinical-grade confidence
- Quick turnaround and low cost for small regions (think 1–2 days, under $150)
- Often overlooked until you hit ambiguous results from NGS or third-gen
But let me be clear—when I tried using Sanger to sequence large genes or whole genomes, it was like trying to swim across an ocean with floaties: slow, expensive, and impractical.
Next-Generation Sequencing (NGS): Not Just “Faster Sanger”
NGS gets hyped as the superhero replacing Sanger—and for some projects, it is—but its power isn’t just speed. It’s about scale through massive parallelization.
Instead of reading DNA one fragment at a time like Sanger, NGS chops your genome into millions of tiny pieces (100–300 base pairs), sequences them all simultaneously, then uses software to stitch them back together. This is why you can get gigabases of data in days instead of years.
Personal blunder alert: During my postdoc cancer genomics work, we relied heavily on NGS panels covering thousands of genes. But early on, I overlooked repetitive regions where NGS’s short reads can’t provide confident maps. That led to missing some important structural variants until I realized short reads have serious blind spots.
Key NGS specifics:
- Depth matters: 30x coverage is typical for human genome WGS to confidently call variants
- Cost per base is way lower than Sanger (think under $0.01 per megabase now)
- Great for broad questions—exomes, whole genomes, targeted panels
Just don’t assume NGS can resolve everything—repeat regions and large insertions/deletions often need another approach.
Third-Generation Sequencing: The Long-Read Game Changer
When I first heard about PacBio and Oxford Nanopore sequencing, I thought they were just shiny new versions of NGS. Nope. They’re fundamentally different beasts.
They sequence single DNA molecules in real time without amplification—meaning they generate ultra-long reads (over 10 kb up to hundreds of kb). This lets you see complex genomic architecture that short-read NGS simply can’t untangle.
I remember collaborating on a cancer project where we suspected large insertions were driving tumor growth but couldn’t confirm it with NGS data alone. Using PacBio SMRT sequencing revealed these structural variants clearly—it felt like finally getting the missing pieces in a jigsaw puzzle after weeks of frustration.
What’s behind the scenes? PacBio watches fluorescent signals as polymerase incorporates bases; Nanopore detects changes in electrical current as DNA passes through tiny pores.
Trade-offs worth knowing:
- Higher raw read error rates (~10-15%) that require consensus building—so you need good depth and bioinformatics skills
- More expensive per base than NGS (roughly 5–10 times) but costs are dropping fast
- Slower throughput but improving steadily
Third-gen shines when your research depends on resolving repeats, complex rearrangements, or full-length haplotypes.
The Comparison We All Wish Was Clear From Day One
| Technology | Read Length | Throughput | Accuracy | Cost per Base | Best Use Case |
|---|---|---|---|---|---|
| Sanger | ~700–900 bp | Low | Very high (>99.99%) | Moderate | Targeted validation of mutations |
| Next-Gen (Illumina) | 100–300 bp | Very high | High (>99%) | Low | Whole/exome genomes; multiplex panels |
| Third-Gen (PacBio/Ox) | >10 kb to >100 kb | Moderate | Moderate raw; improved with consensus | Higher | Structural variants; complex regions |
Mistakes Almost Everyone Makes but No One Talks About
-
Trying to use one tech for everything: When I designed CRISPR guides based on incomplete short-read data alone, no phenotype showed up because some off-target effects were missed—lesson learned: your sequencing method needs to match your biological question precisely.
-
Ignoring how read length influences resolution: Short reads can’t confidently map repetitive regions or structural variants—yet many beginners overlook this until their data looks messy.
-
Underestimating bioinformatics complexity: Each technology produces very different raw data formats requiring dedicated pipelines (Nanopore electrical signals vs Illumina fluorescence). Jumping into data generation without planning analysis resources can lead to wasted money and months lost.
What I Tell Newcomers Starting Out Today
You don’t have to become an expert in all technologies overnight—that overwhelmed feeling is normal and counterproductive.
Focus on mastering the core strengths and weaknesses: start with NGS for broad coverage projects; bring in Third-Generation methods when tackling complex genomic architectures; keep Sanger ready for precise validation checks.
How To Make Your Next Genome Sequencing Choice Without Regret
-
Write down your exact goal: Is it mutation confirmation? Use Sanger. Whole genome at scale? Go NGS. Need long-read insights? Third-gen is your friend.
-
Check recent product updates from Illumina, PacBio, Oxford Nanopore—they innovate fast and pricing shifts frequently.
-
Budget realistically: Sanger costs maybe $100-$200 per assay; Illumina WGS around $600-$1000 per sample depending on coverage; third-gen often runs above $2000 currently but adds unique value.
-
Talk early with bioinformaticians—the right pipelines save months later.
-
Get hands-on experience through workshops or cloud platforms like DNAnexus or Terra before committing big funds.
Genome sequencing isn’t one-size-fits-all technology but a toolkit where choosing the right instrument saves you headaches—and cash—in the long run. Early on, assuming all methods just differ in speed almost doubled my project timelines and confusion. The simple truth? Understand what each tech really does inside your sample’s DNA story before jumping in.
If I could boil it down to one thing: define your biological question clearly first—then pick the sequencing approach tailored to answer that question most reliably and cost-effectively.
That clarity transformed my research—and it will do the same for yours.



